



This is featured post 1 title
Replace these every slider sentences with your featured post descriptions.Go to Blogger edit html and find these sentences.Now replace these with your own descriptions.This theme is Bloggerized by Lasantha - Premiumbloggertemplates.com.

This is featured post 2 title
Replace these every slider sentences with your featured post descriptions.Go to Blogger edit html and find these sentences.Now replace these with your own descriptions.This theme is Bloggerized by Lasantha - Premiumbloggertemplates.com.

This is featured post 3 title
Replace these every slider sentences with your featured post descriptions.Go to Blogger edit html and find these sentences.Now replace these with your own descriptions.This theme is Bloggerized by Lasantha - Premiumbloggertemplates.com.

Monday, December 20, 2010
Saturday, December 18, 2010
User-defined Functions:gcufbioinfo
 2:53 PM
2:53 PM
 GCbioinfo
GCbioinfo
Let's create another function. How about
You can see that the
reverse? >>> def reverse(s):
... """Return the sequence string in reverse order."""
... letters = list(s)
... letters.reverse()
... return ''.join(letters)
...
>>> reverse('CCGGAAGAGCTTACTTAG')
'GATTCATTCGAGAAGGCC' s" instead of "dna". You can name your arguments whatever you like in Python. It is something of a convention to use short names based on their expected value or meaning. So "s" for string is fairly common in Python code. The other reason to use "s" instead of "dna" in this example is that this function works correctly on any string, not just strings representing dna sequences. So "s" is a better reflection of the generic utility of this function than "dna". You can see that the
reverse function takes in a string, creates a list based on the string, and reverses the order of the list. Now we need to put the list back together as a string so we can return a string. Python string objects have a join() method that joins together a list into a string, separating each list element by a string value. Since we do not want any character as a separator, we use the join() method on an empty string, represented by two quotes ('' or "").User-defined Functions:gcufbioinfo
2:29 PM
GCbioinfo
Here is the process for creating your own function in Python. The first line begins with the keyword
Let's define some functions in the PyCrust shell. Then we can try each function with some sample data and see the result returned by the function.

def, is followed by the name of the function and any arguments (expected input values) surrounded by parentheses, and ends with a colon. Subsequent lines make up the body of the function and must be indented. If a string comment appears in the first line of the body, it becomes part of the documentation for the function. The last line of a function returns a result.Let's define some functions in the PyCrust shell. Then we can try each function with some sample data and see the result returned by the function.

>>> def transcribe(dna):
... """Return dna string as rna string."""
... return dna.replace('T', 'U')
...
>>> transcribe('CCGGAAGAGCTTACTTAG')
'CCGGAAGAGCUUACUUAG' Python Functions:gcufbioinfo
1:41 PM
GCbioinfo
Functions perform an operation on one or more values and return a result. Python comes with many pre-defined functions, as well as the ability to define your own functions. Let's look at a couple of the built-in functions:
len() returns the number of items in a sequence; dir() returns a list of strings representing the attributes of an object; list() returns a new list initialized from some other sequence. >>> dna = 'CTGACCACTTTACGAGGTTAGC'
>>> bases = ['A', 'C', 'G', 'T']
>>> len(dna)
22
>>> len(bases)
4
>>> dir(dna)
['__add__', '__class__', '__contains__', '__delattr__',
'__doc__', '__eq__', '__ge__', '__getattribute__', '__getitem__',
'__getslice__', '__gt__', '__hash__', '__init__', '__le__',
'__len__', '__lt__', '__mul__', '__ne__', '__new__', '__reduce__',
'__repr__', '__rmul__', '__setattr__', '__str__', 'capitalize',
'center', 'count', 'decode', 'encode', 'endswith', 'expandtabs',
'find', 'index', 'isalnum', 'isalpha', 'isdigit', 'islower',
'isspace', 'istitle', 'isupper', 'join', 'ljust', 'lower',
'lstrip', 'replace', 'rfind', 'rindex', 'rjust', 'rstrip', 'split',
'splitlines', 'startswith', 'strip', 'swapcase', 'title',
'translate', 'upper']
>>> dir(bases)
['__add__', '__class__', '__contains__', '__delattr__',
'__delitem__', '__delslice__', '__doc__', '__eq__', '__ge__',
'__getattribute__', '__getitem__', '__getslice__', '__gt__',
'__hash__', '__iadd__', '__imul__', '__init__', '__le__', '__len__',
'__lt__', '__mul__', '__ne__', '__new__', '__reduce__', '__repr__',
'__rmul__', '__setattr__', '__setitem__', '__setslice__', '__str__',
'append', 'count', 'extend', 'index', 'insert', 'pop', 'remove',
'reverse', 'sort']
>>> list(dna)
['C', 'T', 'G', 'A', 'C', 'C', 'A', 'C', 'T', 'T', 'T',
'A', 'C', 'G', 'A', 'G', 'G', 'T', 'T', 'A', 'G', 'C'] Python Lists:gcufbioinfo
1:20 PM
GCbioinfo
Where Python strings are limited to characters, Python lists have no limitations. Python lists are ordered sequences of arbitrary Python objects, including other lists. In addition, you can insert, delete and replace elements in a list. Lists are written as a series of objects, separated by commas, inside of square brackets. Let's look at some lists, and some operations you can perform on lists.

>>> bases = ['A', 'C', 'G', 'T']
>>> bases
['A', 'C', 'G', 'T']
>>> bases.append('U')
>>> bases
['A', 'C', 'G', 'T', 'U']
>>> bases.reverse()
>>> bases
['U', 'T', 'G', 'C', 'A']
>>> bases[0]
'U'
>>> bases[1]
'T'
>>> bases.remove('U')
>>> bases
['T', 'G', 'C', 'A']
>>> bases.sort()
>>> bases
['A', 'C', 'G', 'T']In this example we created a list of single characters that we called bases. Then we added an element to the end, reversed the order of all the elements, retrieved elements by their index position, removed an element with the value'U', and sorted the elements. Removing an element from a list illustrates a situation where we need to supply theremove()method with an additional piece of information, namely the value that we want to remove from the list. As you can see in the picture below, PyCrust takes advantage of Python's ability to let us know what is required for most

operations by displaying that information in a call tip pop-up window. Python Strings:gcufbioinfo
1:05 PM
GCbioinfo
'), double (") or triple (''' or """) quotes. In the example we assigned the string literal CTGACCACTTTACGAGGTTAGC to the variable named dna. >>> dna = 'CTGACCACTTTACGAGGTTAGC'>>> dna
'CTGACCACTTTACGAGGTTAGC' .) operator, then the name of the method followed by opening and closing parentheses.>>> dna.lower()
'ctgaccactttacgaggttagc' s[i]. Indexing begins at zero, so s[0] returns the first character in the string, s[1] returns the second, and so on.>>> dna[0]
'C'
>>> dna[1]
'T'
>>> dna[2]
'G'
>>> dna[3]
'A' 
Beginning Python for Bioinformatics:gcufbioinfo
12:36 PM
GCbioinfo
Bioinformatics, the use of computers in biological research, is the newest wrinkle on one of the oldest pursuits--trying to uncover the secret of life. While we may not know all of life's secrets, at the very least computers are helping us understand many of the biological processes that take place inside of living things. In fact, the use of computers in biological research has risen to such a degree that computer programming has now become an important and almost essential skill for today's biologists.
The purpose of this article is to introduce Python as a useful and viable development language for the computer programming needs of the bioinformatics community. In this introduction, we'll identify some of the advantages of using Python for bioinformatics. Then we'll create and demonstrate examples of working code to get you started. In subsequent articles we'll explore some significant bioinformatics projects that make use of Python.
What I concluded:
6:57 AM
GCbioinfo
Conclusion
From all these research papers I have concluded that from all the data mining techniques PCA is considered to be the best because it reduces large data set into smaller ones and narrow down our research that we can useful information from small data sets. Dimension of a large dataset can be reduced by using principal component analysis which is considered as one of the most popular and useful statistical method. This method transforms the original data in to new dimensions.
Predicting Breast Cancer Survivability Using Data Mining Techniques
6:55 AM
GCbioinfo
Last edited by
Quratt ul ain Siddique
Summary:
This paper presents the prediction of the survivability rate of cancer using data mining techniques. In this paper scientists investigated three data mining like Naïve Bayes, the back-propagated neural network, and the C4.5 decision tree algorithms. C4.5 decision tree algorithm is considered to best from remaining two methods. In this study SEER data is used and introduced a pre-classification approach that take into account three variables: Survival Time Recode (STR), Vital Status Recode (VSR), and Cause of Death (COD).
In this paper three data mining techniques are used to find which one is best to find breast cancer survivability rate. In this research Weka toolkit is used for experimentation that used three data mining algorithms in this research raw SEER data is get before processing using different tools.
In this approach missing information from SEER data is completely ignored and included three approaches like STR, VSR, COD. In this study three data mining techniques is compared. The goal is to attain high precision and accuracy from these techniques. These are actually the matrices which are mostly used for the retrieval of the information but there they are considered to be related to the other existing metrics such as specificity and sensitivity.
This paper discussed and resolved the issues, algorithms and techniques and problems related to predict Breast Cancer Survivability using SEER database. It also discussed that among three data minig techniques the C4.5 decision tree is considered to be the best because it shown maximum accuracy ,precision and recall metrics.
Combined Supervised and Unsupervised Learning in Genomic Data Mining
6:50 AM
GCbioinfo
Last edited by
Quratt ul ain Siddique
Summary:
In this paper they introduced the most comprehensive method for predicting the function of proteins. Their approach differs in several respects from the earlier work in that it uses a multistage decomposition that makes use of both unsupervised and supervised machine learning techniques; they refer to this as Unsupervised-Supervised Tree (UST) algorithm.
The typical first stage (optional) of the UST uses clustering algorithms such as neural network self organizing maps (SOMs) and K-means; this is the unsupervised stage. Subsequent indispensable stages typically involve constructing a Maximum Contrast Tree (MCT) so that protein functional relationships can be mapped onto the relational tree structure.
The MCTs are a family of completely independent algorithms that can be used alone. Testing is based on a newly developed MLIC (Multiple-Labeled Instance Classifier) based on supervised K nearest neighbor classifier on the tree structure. Performance has been compared with the decision tree C4.5 and C5 programs and with support vector machines.
Based on the experiments, UST algorithms appear to perform considerably better than decision tree algorithms C4.5 and C5, and support vector machines, and can provide a viable alternative to supervised or unsupervised methods alone. In addition, UST and MLIC classifiers are capable of handling protein functional classes with a small number of proteins (rare events), and also handle multifunctional proteins. The abilities of the USTs and MLICs to handle such cases means that a larger dataset can be used, which may provide deeper insight into protein functional relationships at the genomic level, and thus may lead to a better understanding of evolution at a molecular and genomic level.
Acute Coronary Syndrome Prediction Using Data Mining Techniques- An Application
6:45 AM
GCbioinfo
Last edited by
Quratt ul ain Siddique
Summary:
In this research paper data mining techniques are used to investigate the factors that are responsible for enhancing the risk of acute coronary syndrome. They have applied binary regression to factors that effecting the dependent variable. For the better performance of regression model in predicting coronary syndrome the reduction technique which is principle component analysis is used and applied. Based on results of data reduction, they have considered only 14 out of sixteen factors.
In this research paper logistic regression model is used to find the factors which are responsible for this Acute Coronary Syndrome (ACS). For the analysis of this problem data mining technique is used for the comparasion of the persons who have ACS or who don’t have.
In this paper first data reduction techniques are applied that reduce the dimensions. After data reduction, the fourteen independent variables are age, gender, smoke, hypertension, family history, diabetics mellitus, fasting blood sugar, random blood sugar, cholesterol, streptokinase, blood pressure (systolic), blood pressure (diastolic), heart rate and pulse rate. After the calculation of corresponding significance of smoking which is “0” indicating that it has a high prevalence in the risk of ACS. The calculation of wald statistics indicates positive coefficients of HR, RBS, and BPS revealed that the risk of ACS increases with the increasing value of these factors.
The negative coefficients of BPd and PR revealed that the more the negative these values the more the increase in the risk of this disease. They observed that smoking is considered to be the worst cause of this Acute Coronary Sysndrome.
Biological Data Mining for Genomic Clustering Using Unsupervised Neural Learning:
6:40 AM
GCbioinfo
Last edited by
Quratt ul ain Siddique
Quratt ul ain Siddique
Summary:
Among the well-known techniques of DNA-string matching are the Smith-Waterman algorithm, for local alignment, the Needleman-Wunsch algorithm for global alignment, Hidden Markov’s model, matrix model, evolutionary algorithms for multiple sequence alignment etc. These works, though extremely valuable, have their limitations.
Principal Component analysis is then employed on the DNA-descriptors for N sampled instances. The principal component analysis yields a unique feature descriptor for identifying the species from its genome sequence. The variance of the descriptors for a given genome sequence being negligible, the proposed scheme finds extensive applications in automatic species identification. PCA is actually used to find the structural signature within a sequence or species which is used to differentiate the specie without the loss of accuracy. Since PCA is a well-known tool for data reduction without loss of accuracy, we claim that our results on feature extraction from the genome database are also free from loss of accuracy.
It is quite evident that the feature descriptors provide a more unique identifier for the species from its genomic data. Thus we have certainly gained an advantage by incorporating the data reduction tool PCA into our search for an effective identifier for a species. It has been found out that the DNA-descriptors obtained from different samples of the same species contain wide disparities. But the Feature Descriptors obtained after processing a different set of DNA-descriptors are unique and present absolutely no significant disparities. Hence the Feature Descriptor Diagrams can be used as the unique representation of the genomic characteristics of the different species.
Feature Discriptors are more accurate in identifying different because they are obtained from Mitochondrial genome not from the whole genome sequence and by applying PCA on them gave accurate results.
If only the frequency count is plotted then we do get some difference from species to species but it is not enough to distinguish between them. This is where PCA comes in. When we applied PCA to data to get Features Descriptors Diagrams of different species then we are able to differentiate species. Moreover when feature descriptor vectors for similar species are calculated, they are effective in bringing out the similarities in the species though they still retain their individual distinguishing features. By constructing the Feature Descriptor Diagram for the species we get best identifier for the particular specie.
An alternative approach to automatic species classification and identification of species using Self-Organizing Feature Map is also discussed in the paper. The computational map is trained by using the DNA-descriptors from different species as the training inputs. The maps for different dimensions are constructed and analyzed for optimum performance. The scheme presents a novel method for identifying a species from its genome sequence with the help of a two dimensional map of neuronal clusters, where each cluster represents a particular species. The map is shown to provide an easier technique for recognition and classification of a species based on its genomic data. Maps of different dimensions are constructed and analyzed on the basis of their efficiency in clustering the extracted features from genomic data of different species.
Also the SOFM can help us demonstrate homology between new sequences and existing phyla. When a new sequence is obtained then its DNA Descriptor is computed and distance between existing neurons is calculated. Then winning neuron from which it has very low distance declares to which species it belongs or it belongs to a new specie. Then to which phylum this specie belongs.
Currently, works in Bioinformatics and biological data mining are aimed at discovering the parts of the DNA sequence that are translated to proteins and to which functions they are involved in forming different parts of the body, ie to identify the genes and their functionality. Another trend to predict structure and functions of these sequences. But the novelity of this work is automatic species identification from genomic data.
Data Mining and Visualization of Mouse Genome Data:
6:35 AM
GCbioinfo
Last edited by
Quratt ul ain Siddique
Quratt ul ain Siddique
Summary:
This paper discusses the data mining of the genomics of the mouse that is an area of importance because of its relationship to understanding of basic genetics of other mammals and in particular the human as well as livestock genetics and its breeding.
The data mining tools of multiplot, data partition, clustering, self-organized maps (SOM), regression, association, and neural networks were all used in this research The paper has demonstrated the data mining and visualization results including virtual gene map, mouse genomic features on chromosome, clustering, cluster proximity, T-Scores effect, self-organizing map, and regression analysis. One of the novelties of this research is that the data mining is performed at the genomic level of a mammal that is commonly used as prototype testings for humans.
The data mining performed on the mouse genome data indicated a linearity of regression for the B16F0 Chromosone, significant reduction in the average error upon using neural network algorithms, significant effect in the visulization plots upon using self-organized maps (SOM), and a nonlinear relationship of the cubicclustering criterion with discontinities when the number of clusters reached 22 and 38.
The results of data mining performed also indicated that it was useful to visualize at the genomic level for the mouse data. The analysis shown here can also help researchers who are interested in genome data, and others to visualize the use of data mining at this micro dimensional level.
Future directions of the research are to continue to perform more data mining of the mouse genome data. This may entail using other data mining tools and software. Other future directions are to perform data mining for other data bases such as for other mammals that are of evolutionary relationship to humans, and also other genomic databases of differing dimensionalities to contrast the findings of the research presented in this paper.
Saturday, November 27, 2010
Rasmol:gcufbioinfo
10:16 AM
GCbioinfo
Click on different balls of your molecule and it will give information about the particular atom you clicked in which it exists.


rasmol:gcufbioinfo
10:10 AM
GCbioinfo
open raswin and select file>information to get complete information about the particular molecule.


Rasmol:gcufbioinfo
10:01 AM
GCbioinfo
Select raswin from your list and open it select file>open and select file from any derive and open it into rasmol .
click Display>and select different options to view your molecule.

click Display>and select different options to view your molecule.

Rasmol:gcufbioinfo
9:46 AM
GCbioinfo
This was the way in which you can visualize the molecule using Rasmol on the pdb page but if you want to view the saved page of pdb so click the option "Display files".
And save this file to any directory with file format ".pdb" so now you can visualize already saved molecule.
And save this file to any directory with file format ".pdb" so now you can visualize already saved molecule.
Rasmol:gcufbioinfo
9:34 AM
GCbioinfo
To view Structure using Rasmol you must have pdb file of your macromolecule and micromolecule so go to PDB
enter protein name for which you want to view the structure. As I entered DNA Polymerase so the result is http://www.pdb.org/pdb/explore/explore.do?structureId=3OGU
the out put is as:
 At the right of this page you can there is the option of Download files so click this. After clicking this select "PDB files(gz)".then download and install winzip to your system and download this file automatically to raswin and view the structure in rasmol.
At the right of this page you can there is the option of Download files so click this. After clicking this select "PDB files(gz)".then download and install winzip to your system and download this file automatically to raswin and view the structure in rasmol.
enter protein name for which you want to view the structure. As I entered DNA Polymerase so the result is http://www.pdb.org/pdb/explore/explore.do?structureId=3OGU
the out put is as:

What kinds of molecules can RasMol display?:Gcufbioinfo
9:25 AM
GCbioinfo
RasMol can display any molecule for which a 3-dimensional structure is available. 3D structures have not been determined for many molecules of great interest; these, RasMol cannot display.
In order to display a molecule, RasMol needs a data file called an atomic coordinate file. This data file specifies the position of every atom in the molecule, as cartesian coordinates X, Y, and Z.
Three-dimensional structures can be predicted for many small molecules, but must be determined empirically for macromolecules. The most common method for determining structure is X-ray diffraction analysis of a crystal. Nuclear magnetic resonance (NMR) can also be used. Some structures are available only as theoretical models, often based on related molecules for which empirical structures have been determined.
In order to display a molecule, RasMol needs a data file called an atomic coordinate file. This data file specifies the position of every atom in the molecule, as cartesian coordinates X, Y, and Z.
Three-dimensional structures can be predicted for many small molecules, but must be determined empirically for macromolecules. The most common method for determining structure is X-ray diffraction analysis of a crystal. Nuclear magnetic resonance (NMR) can also be used. Some structures are available only as theoretical models, often based on related molecules for which empirical structures have been determined.
Rasmol Tool:
8:19 AM
GCbioinfo
RasMol is a computer program written for molecular graphics visualization intended and used primarily for the depiction and exploration of biological macromolecule structures, such as those found in the Protein Data Bank. It was originally developed by Roger Sayle in the early 90s get started this useful tool Download Rasmol
here. Install Rasmol to your system and get started.
here. Install Rasmol to your system and get started.
Thursday, November 25, 2010
HTML Calculator:gcufbioinfo
8:25 PM
GCbioinfo
paste this code to your source code of your calculator in head part
function sqrt(form)
{
form.displayvalue.value=Math.sqrt(form.displayvalue.value);
}
function sq(form)
{
form.displayvalue.value=eval(form.displayvalue.value)*eval(form.displayvalue.value);
}
function ln(form)
{
form.displayvalue.value=Math.exp(form.displayvalue.value);
}
function equal(form)
{
form.displayvalue.value=eval(form.displayvalue.value);
}
function delChar(input)
{
input.value=input.value.substring(0,input.value-length-1);
}
function addChar(input,character)
{
if(input.value==null||input.value=="0")
input.value=character
else
input.value+=character
}
Insha Allah you will see your functioning very soon regards Admin Quratt ul ain Siddique.
function sqrt(form)
{
form.displayvalue.value=Math.sqrt(form.displayvalue.value);
}
function sq(form)
{
form.displayvalue.value=eval(form.displayvalue.value)*eval(form.displayvalue.value);
}
function ln(form)
{
form.displayvalue.value=Math.exp(form.displayvalue.value);
}
function equal(form)
{
form.displayvalue.value=eval(form.displayvalue.value);
}
function delChar(input)
{
input.value=input.value.substring(0,input.value-length-1);
}
function addChar(input,character)
{
if(input.value==null||input.value=="0")
input.value=character
else
input.value+=character
}
Insha Allah you will see your functioning very soon regards Admin Quratt ul ain Siddique.
Monday, November 22, 2010
HTML Calculator:gcufbioinfo
8:53 AM
GCbioinfo
Add this code to your code of calculator in head portion
<head>
<script type="text/javascript">
function cos(form)
{
form.displayvalue.value=Math.cos(form.displayvalue.value);
}
function sin(form)
{
form.displayvalue.value=Math.sin(form.displayvalue.value);
}
function tan(form)
{
form.displayvalue.value=Math.tan(form.displayvalue.value);
}
</script>
</head>
I will tell you other codes as soon as possible and you will see Insha Allah your calculator functional
<head>
<script type="text/javascript">
function cos(form)
{
form.displayvalue.value=Math.cos(form.displayvalue.value);
}
function sin(form)
{
form.displayvalue.value=Math.sin(form.displayvalue.value);
}
function tan(form)
{
form.displayvalue.value=Math.tan(form.displayvalue.value);
}
</script>
</head>
I will tell you other codes as soon as possible and you will see Insha Allah your calculator functional
HTML Calculator:gcufbioinfo
8:33 AM
GCbioinfo
As I told you how to generate calculator using html codes so this is again calculator with some changes so plz try it. I will tell you how to make it functional
<html>
<title>Calculator</title>
<head></head>
<body>
<table border="2">
<tr>
<td><input name="displayvalue" size="39" value="0"></td>
<tr>
<td>
<table border="2">
<tr>
<td>
<input type="button" value=" char ">
</td>
<td>
<input type="button" value=" delete ">
</td>
<td>
<input type="button" value=" = ">
</td>
</tr>
</table>
</td>
<tr>
<td>
<table border="2">
<tr>
<td>
<input type="button" value=" exp ">
</td>
<td>
<input type="button" value=" 7 ">
</td>
<td>
<input type="button" value=" 8 ">
</td>
<td>
<input type="button" value=" 9 ">
</td>
<td>
<input type="button" value=" / ">
</td>
</tr>
<tr>
<td>
<input type="button" value=" ln ">
</td>
<td>
<input type="button" value=" 4 ">
</td>
<td>
<input type="button" value=" 5 ">
</td>
<td>
<input type="button" value=" 6 ">
</td>
<td>
<input type="button" value=" * ">
</td>
</tr>
<tr>
<td>
<input type="button" value=" sqrt ">
</td>
<td>
<input type="button" value=" 1 ">
</td>
<td>
<input type="button" value=" 2 ">
</td>
<td>
<input type="button" value=" 3 ">
</td>
<td>
<input type="button" value=" - ">
</td>
</tr>
<tr>
<td>
<input type="button" value=" sq ">
</td>
<td>
<input type="button" value=" 0 ">
</td>
<td>
<input type="button" value=" . ">
</td>
<td>
<input type="button" value=" % ">
</td>
<td>
<input type="button" value=" + ">
</td>
</tr>
<tr>
<td>
<input type="button" value=" ( ">
</td>
<td>
<input type="button" value=" cos ">
</td>
<td>
<input type="button" value=" sin ">
</td>
<td>
<input type="button" value=" tan ">
</td>
<td>
<input type="button" value=" ) ">
</td>
</tr>
</table>
</td>
</tr>
</tr>
</tr>
</table>
</body>
</html>
out put is as:
 Now I tell you how to make it functional
Now I tell you how to make it functional
Quratt ul ain Siddique
<html>
<title>Calculator</title>
<head></head>
<body>
<table border="2">
<tr>
<td><input name="displayvalue" size="39" value="0"></td>
<tr>
<td>
<table border="2">
<tr>
<td>
<input type="button" value=" char ">
</td>
<td>
<input type="button" value=" delete ">
</td>
<td>
<input type="button" value=" = ">
</td>
</tr>
</table>
</td>
<tr>
<td>
<table border="2">
<tr>
<td>
<input type="button" value=" exp ">
</td>
<td>
<input type="button" value=" 7 ">
</td>
<td>
<input type="button" value=" 8 ">
</td>
<td>
<input type="button" value=" 9 ">
</td>
<td>
<input type="button" value=" / ">
</td>
</tr>
<tr>
<td>
<input type="button" value=" ln ">
</td>
<td>
<input type="button" value=" 4 ">
</td>
<td>
<input type="button" value=" 5 ">
</td>
<td>
<input type="button" value=" 6 ">
</td>
<td>
<input type="button" value=" * ">
</td>
</tr>
<tr>
<td>
<input type="button" value=" sqrt ">
</td>
<td>
<input type="button" value=" 1 ">
</td>
<td>
<input type="button" value=" 2 ">
</td>
<td>
<input type="button" value=" 3 ">
</td>
<td>
<input type="button" value=" - ">
</td>
</tr>
<tr>
<td>
<input type="button" value=" sq ">
</td>
<td>
<input type="button" value=" 0 ">
</td>
<td>
<input type="button" value=" . ">
</td>
<td>
<input type="button" value=" % ">
</td>
<td>
<input type="button" value=" + ">
</td>
</tr>
<tr>
<td>
<input type="button" value=" ( ">
</td>
<td>
<input type="button" value=" cos ">
</td>
<td>
<input type="button" value=" sin ">
</td>
<td>
<input type="button" value=" tan ">
</td>
<td>
<input type="button" value=" ) ">
</td>
</tr>
</table>
</td>
</tr>
</tr>
</tr>
</table>
</body>
</html>
out put is as:

Quratt ul ain Siddique
Sunday, November 21, 2010
imageJ:gcufbioinfo
5:57 AM
GCbioinfo
Open your image in imageJ and select adjust in imageJ adjust>color and contrast you can use these sliders to adjust display range, either down from the top or up from the bottom.
Medical Image Processing tool:
5:46 AM
GCbioinfo
Now we are going to see how to view an image using imageJ on gcufbioinfo.
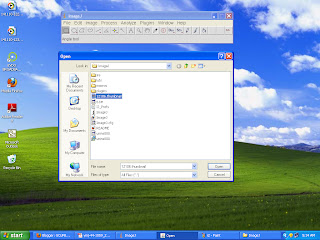
First save an image especially a medical image to your system and copy this image and go to C derive then program files and then imagej folder and paste this image into it. then open imageJ from start menu and click file >open as shown in gcufbioinfo and select the picture to view.
 you can view your image using this tool .
you can view your image using this tool .
First save an image especially a medical image to your system and copy this image and go to C derive then program files and then imagej folder and paste this image into it. then open imageJ from start menu and click file >open as shown in gcufbioinfo and select the picture to view.

imageJ:gcufbioinfo
5:10 AM
GCbioinfo
You can access imageJ by clicking on start button and click imageJ to click the microscope icon. The out put you can see at gcufbioinfo and click help to get information about imageJ.


Download ImageJ:
4:50 AM
GCbioinfo
To download imageJ first go to the http://rsbweb.nih.gov/ij/
the page will open
 click download
click download
http://rsbweb.nih.gov/ij/download.html
then get latest version of imageJ for your OS . If you are new to use ImageJ then Download ImageJ 1.43 bundled with 32-bit Java 1.6.0_10 (25MB) other wise download without java for windows OS.
the page will open

http://rsbweb.nih.gov/ij/download.html
then get latest version of imageJ for your OS . If you are new to use ImageJ then Download ImageJ 1.43 bundled with 32-bit Java 1.6.0_10 (25MB) other wise download without java for windows OS.
Medical Image Processing tool:
4:37 AM
GCbioinfo
ImageJ:
ImageJ is a public domain, Java-based image processing program developed at the National Institutes of Health. ImageJ was designed with an open architecture that provides extensibility via Java plugins and recordable macros. Custom acquisition, analysis and processing plugins can be developed using ImageJ's built-in editor and a Java compiler. User-written plugins make it possible to solve many image processing and analysis problems, from three-dimensional live-cell imaging, to radiological image processing,multiple imaging system data comparisonsto automated hematology systems.ImageJ's plugin architecture and built in development environment has made it a popular platform for teaching image processing.
ImageJ is a public domain, Java-based image processing program developed at the National Institutes of Health. ImageJ was designed with an open architecture that provides extensibility via Java plugins and recordable macros. Custom acquisition, analysis and processing plugins can be developed using ImageJ's built-in editor and a Java compiler. User-written plugins make it possible to solve many image processing and analysis problems, from three-dimensional live-cell imaging, to radiological image processing,multiple imaging system data comparisonsto automated hematology systems.ImageJ's plugin architecture and built in development environment has made it a popular platform for teaching image processing.
Tuesday, November 2, 2010
primary structure analysis:
8:11 AM
GCbioinfo
Tool:
Radar:De novo repeat detection in protein sequences
I entered the protein albumin in uniprot and get the following result It gave the number of repeats present in the sequence.
http://www.ebi.ac.uk/Tools/es/cgi-bin/jobresults.cgi/radar/radar-20101102-1505421645.html

Radar:De novo repeat detection in protein sequences
I entered the protein albumin in uniprot and get the following result It gave the number of repeats present in the sequence.
http://www.ebi.ac.uk/Tools/es/cgi-bin/jobresults.cgi/radar/radar-20101102-1505421645.html

Monday, November 1, 2010
primary structure analysis
7:49 AM
GCbioinfo
Tools:
Radar :De novo repeat detection in protein sequences go to http://www.ebi.ac.uk/Tools/Radar/index.html
Radar :De novo repeat detection in protein sequences go to http://www.ebi.ac.uk/Tools/Radar/index.html
primary structure analysis:
7:39 AM
GCbioinfo
Tools:
HeliQuest A web server to screen sequences with specific alpha-helical properties HeliQuest calculates from an α-helix sequence its physicochemical properties and amino acid composition and uses the results to screen any databank in order to identify protein segments possessing similar features.
The server is divided into 2 interconnected modules: the sequence analysis module and the screening module.
In addition, the mutation module, (available from the sequence analysis module), allows user to mutate helices manually or automatically by genetic algorithm to create analogues with specific properties.-http://heliquest.ipmc.cnrs.fr/
 After entering the helix sequence of the protein this protein sequence is get by entering the name of the protein in uniprot then paste it by clicking the analysis button and get the following
After entering the helix sequence of the protein this protein sequence is get by entering the name of the protein in uniprot then paste it by clicking the analysis button and get the following
http://heliquest.ipmc.cnrs.fr/cgi-bin/ComputParams.py

HeliQuest A web server to screen sequences with specific alpha-helical properties HeliQuest calculates from an α-helix sequence its physicochemical properties and amino acid composition and uses the results to screen any databank in order to identify protein segments possessing similar features.
The server is divided into 2 interconnected modules: the sequence analysis module and the screening module.
In addition, the mutation module, (available from the sequence analysis module), allows user to mutate helices manually or automatically by genetic algorithm to create analogues with specific properties.-http://heliquest.ipmc.cnrs.fr/

http://heliquest.ipmc.cnrs.fr/cgi-bin/ComputParams.py
 7:19 AM
GCbioinfo
7:19 AM
GCbioinfo
Tools:
ScanSite pI/Mw:Compute the theoretical pI and Mw, and multiple phosphorylation states. Enter a protein name and sequence below. The molecular weight and isoelectric point of this sequence and multiple phosphorylation states will be displayed. http://scansite.mit.edu/calc_mw_pi.html
out put will be

ScanSite pI/Mw:Compute the theoretical pI and Mw, and multiple phosphorylation states. Enter a protein name and sequence below. The molecular weight and isoelectric point of this sequence and multiple phosphorylation states will be displayed. http://scansite.mit.edu/calc_mw_pi.html
out put will be

primary structure analysis 3:
7:06 AM
GCbioinfo
Tool:
Compute pI/Mw: Compute the theoretical isoelectric point (pI) and molecular weight (Mw) from a UniProt Knowledgebase entry or for a user sequence
now go to this tool and get the required result Please enter one or more UniProtKB/Swiss-Prot protein identifiers (ID) (e.g. ALBU_HUMAN) or UniProt Knowledgebase accession numbers (AC) (e.g. P04406), separated by spaces, tabs or newlines. Alternatively, enter a protein sequence in single letter code. The theoretical pI and Mw (molecular weight) will then be computed. previosly I entered the amino acid sequence and now I am going to enter the uniprot AC#.http://www.expasy.org/cgi-bin/pi_tool
it will give you this format of result so click on the following things according to the instructions mentioned Please select one of the following features by clicking on a pair of endpoints, and the computation will be carried out for the corresponding sequence fragment.

Compute pI/Mw: Compute the theoretical isoelectric point (pI) and molecular weight (Mw) from a UniProt Knowledgebase entry or for a user sequence
now go to this tool and get the required result Please enter one or more UniProtKB/Swiss-Prot protein identifiers (ID) (e.g. ALBU_HUMAN) or UniProt Knowledgebase accession numbers (AC) (e.g. P04406), separated by spaces, tabs or newlines. Alternatively, enter a protein sequence in single letter code. The theoretical pI and Mw (molecular weight) will then be computed. previosly I entered the amino acid sequence and now I am going to enter the uniprot AC#.http://www.expasy.org/cgi-bin/pi_tool
it will give you this format of result so click on the following things according to the instructions mentioned Please select one of the following features by clicking on a pair of endpoints, and the computation will be carried out for the corresponding sequence fragment.

Primary structure analysis 2:
6:51 AM
GCbioinfo
Tool :
ProtParam :Physico-chemical parameters of a protein sequence (amino-acid and atomic compositions, isoelectric point, extinction coefficient, etc.)
First go to this page http://www.expasy.org/tools/ select Primary structure analysis and select the first tool as I mentioned. click the tool and I entered the amino acid sequence of protein transgelin-2 in http://www.expasy.org/tools/protparam.html then get the out put page
http://www.expasy.org/cgi-bin/protparam
the out put is

ProtParam :Physico-chemical parameters of a protein sequence (amino-acid and atomic compositions, isoelectric point, extinction coefficient, etc.)
First go to this page http://www.expasy.org/tools/ select Primary structure analysis and select the first tool as I mentioned. click the tool and I entered the amino acid sequence of protein transgelin-2 in http://www.expasy.org/tools/protparam.html then get the out put page
http://www.expasy.org/cgi-bin/protparam
the out put is

Primary structure analysis:
6:36 AM
GCbioinfo
Now we are going to the expasy server for the Primary structure analysis of proteins by using different tools available at expasy server.
I will breifly tell you how to use these tools.
I will breifly tell you how to use these tools.
HTML Calculator:
6:22 AM
GCbioinfo
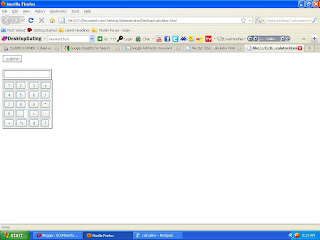
As I told you how to generate a table in html today I am going to develop a calculator by using html table tags but the new things is the button so the code for the button is
<input type="button" value="submit">
<html>
<body>
<input type="button" value="submit">
<br><br>
<table border="2">
<tr><td><input type="text"></td></tr>
<tr><table border="2"><tr><td><input type="button" value="1"></td><td><input type="button" value="2"></td><td><input type="button" value="3"></td><td><input type="button" value="c"></td></tr><tr><td><input type="button" value="4"></td><td><input type="button" value="5"></td><td><input type="button" value="6"></td><td><input type="button" value="/"></td></tr><tr><td><input type="button" value="7"></td><td><input type="button" value="8"></td><td><input type="button" value="9"></td><td><input type="button" value="*"></td></tr><tr><td><input type="button" value="0"></td><td><input type="button" value="."></td><td><input type="button" value="="></td><td><input type="button" value="-"></td></tr><tr><td><input type="button" value="+"></td><td><input type="button" value="%"></td><td><input type="button" value="d"></td><td><input type="button" value="f"></td></tr></table></tr>
</table>
</body>
</html>
<input type="button" value="submit">
<html>
<body>
<input type="button" value="submit">
<br><br>
<table border="2">
<tr><td><input type="text"></td></tr>
<tr><table border="2"><tr><td><input type="button" value="1"></td><td><input type="button" value="2"></td><td><input type="button" value="3"></td><td><input type="button" value="c"></td></tr><tr><td><input type="button" value="4"></td><td><input type="button" value="5"></td><td><input type="button" value="6"></td><td><input type="button" value="/"></td></tr><tr><td><input type="button" value="7"></td><td><input type="button" value="8"></td><td><input type="button" value="9"></td><td><input type="button" value="*"></td></tr><tr><td><input type="button" value="0"></td><td><input type="button" value="."></td><td><input type="button" value="="></td><td><input type="button" value="-"></td></tr><tr><td><input type="button" value="+"></td><td><input type="button" value="%"></td><td><input type="button" value="d"></td><td><input type="button" value="f"></td></tr></table></tr>
</table>
</body>
</html>

this is not a functioning calculator it is just a sekeleton of calculator the functioning of this calculator I will told you latter
regards Admin Quratt ul ain Siddique
Saturday, October 30, 2010
Muliple Sequence Alignment
10:09 AM
GCbioinfo
go to this page to align your sequences using ClustalW paste all of your sequences or upload your text file to get your alignment result
http://www.ebi.ac.uk/Tools/es/cgi-bin/clustalw2/result.cgi?tool=clustalw2&jobid=clustalw2-20101030-1758594671&poll=yes
 This is the multiple sequence alignment of the selected sequences
This is the multiple sequence alignment of the selected sequences
http://www.ebi.ac.uk/Tools/es/cgi-bin/clustalw2/result.cgi?tool=clustalw2&jobid=clustalw2-20101030-1758594671&poll=yes

Muliple Sequence Alignment
9:55 AM
GCbioinfo
For Multiple sequence Alignment you need Fasta sequence for this go to ncbi for nucleotide or at expasy server for protein sequence but now I am going to work with proteins so go to www.expasy.org
enter your query for which you want to retrieve the sequences
or you can also go to uniprot which in my opinion is easy to use and get fasta sequence so go to uniprot and enter protein hemoglobin and get different entries of this protein click them and get fasta sequence like this
 and select many proteins and get their fasta formats like this
and select many proteins and get their fasta formats like this
click on fasta and http://www.uniprot.org/uniprot/P68871.fasta
copy and paste these fasta sequences of many proteins in a text file and collect all these sequences in a text document.
In this format I pasted my all sequences in my notepad

enter your query for which you want to retrieve the sequences
or you can also go to uniprot which in my opinion is easy to use and get fasta sequence so go to uniprot and enter protein hemoglobin and get different entries of this protein click them and get fasta sequence like this

click on fasta and http://www.uniprot.org/uniprot/P68871.fasta
copy and paste these fasta sequences of many proteins in a text file and collect all these sequences in a text document.
In this format I pasted my all sequences in my notepad

ClustalW:
9:27 AM
GCbioinfo
ClustalW is a general purpose multiple sequence alignment program for DNA and proteins it produces biologically meaning full multiple sequence alignments of divergent sequences it calculates the best match for selected sequences that lines up so that the identities and similarities and differences and evolutionary relationships can be seen via viewing cladograms or phylograms.
PDB structures part 3
8:27 AM
GCbioinfo
The previous page contains the PDB ID of the particular protein
http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid=34605
get this PDB ID and entered into PDB and get the same 3D structure of the protein
http://www.rcsb.org/pdb/explore/explore.do?structureId=1WYM

http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid=34605
get this PDB ID and entered into PDB and get the same 3D structure of the protein
http://www.rcsb.org/pdb/explore/explore.do?structureId=1WYM

PDB structures part 2
8:18 AM
GCbioinfo
As we entered the gene name to ncbi and get complete information about the gene and also get the protein which is encoded by the gene. Get this name and entered into the structure database in ncbi. http://www.ncbi.nlm.nih.gov/sites/structure/?term=Transgelin-2
 the protein entered is Transgelin-2
the protein entered is Transgelin-2
http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid=34605
view 3D structure using cn3d which is a structure viewer I mentioned in my very previous tutorial how to use and download cn3d 4.0 to your systems


http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid=34605
view 3D structure using cn3d which is a structure viewer I mentioned in my very previous tutorial how to use and download cn3d 4.0 to your systems

PDB Structures
6:30 AM
GCbioinfo
As you know PDB is used for finding the structure of different proteins for this I have selected different genes and get their proteins you can see.
I select three Tumor Supressor Genes
1.

I select three Tumor Supressor Genes
1.
p16INK4a
2.
p53
3.
RB - the retinoblastoma gene
entered these genes in ncbi http://www.ncbi.nlm.nih.gov/
select nucleotide in search and enter the gene name
p16INK4a get this page http://www.ncbi.nlm.nih.gov/nuccore/NM_001001522.1

3.PDB:
4:27 AM
GCbioinfo
PDB is a protein data bank it contains information about the structure and function of the reported proteins. Not only the information about the protein is present but also of many macromolecules like nucleic acids and carbohydrates.
How to Search PDB:
go to http://www.rcsb.org/pdb/explore/motm.do
enter PDB ID if you don't know the ID then enter the text methylenetetrahydrofolate reductase to get the structural information of that protein
I entered this text then get this result go to this page http://www.rcsb.org/pdb/results/results.do?outformat=&qrid=FA02BFE5&tabtoshow=Current

How to Search PDB:
go to http://www.rcsb.org/pdb/explore/motm.do
enter PDB ID if you don't know the ID then enter the text methylenetetrahydrofolate reductase to get the structural information of that protein
I entered this text then get this result go to this page http://www.rcsb.org/pdb/results/results.do?outformat=&qrid=FA02BFE5&tabtoshow=Current

Friday, October 29, 2010
2. PUBCHEM:
4:43 AM
GCbioinfo
http://pubchem.ncbi.nlm.nih.gov/
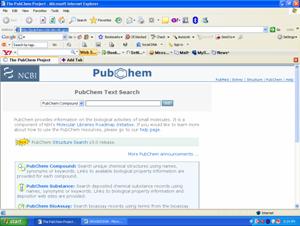
if you enter chlorpropamide which is the molecule use for the treatment of Type II diebetes it will give complete information about the structure and function of the molecule.
 PubChem provides information on the biological activities of small molecules. It is a component of NIH's Molecular Libraries Roadmap Initiative.
PubChem provides information on the biological activities of small molecules. It is a component of NIH's Molecular Libraries Roadmap Initiative.
visit this page I have entered the molecule name
http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=2727&loc=ec_rcs

if you enter chlorpropamide which is the molecule use for the treatment of Type II diebetes it will give complete information about the structure and function of the molecule.
visit this page I have entered the molecule name
http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=2727&loc=ec_rcs

Out put:
3:57 AM
GCbioinfo
I entered the disorder diabetes melitis
and it gives the results that you get by accessing this page
http://www.drugbank.ca/drugs/DB00672

and it gives the results that you get by accessing this page
http://www.drugbank.ca/drugs/DB00672

1. LIGAND PREPARATION:
3:46 AM
GCbioinfo
Ligand preparation further divided into different heading namely, ligand retrieval or collection, liand conversion and ligand analysis
a. Ligands Collection:
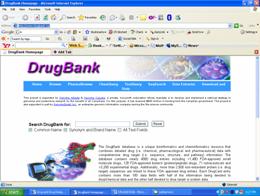
1. DRUGBANK :http://www.drugbank.ca/
first go to this link enter your protein or condition like any disease and disorder it will give you complete information about the drug which is designed for the particular purpose eg enter anxiety disorder in drugbank it will give you different ligands of that particular disorder and protein and also gives complete information about the target protein.
The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. The database contains nearly 4800 drug entries including >1,480 FDA-approved small molecule drugs, 128 FDA-approved biotech (protein/peptide) drugs, 71 nutraceuticals and >3,200 experimental drugs. Additionally, more than 2,500 non-redundant protein (i.e. drug target) sequences are linked to these FDA approved drug entries. Each DrugCard entry contains more than 100 data fields with half of the information being devoted to drug/chemical data and the other half devoted to drug target or protein data.

a. Ligands Collection:
1. DRUGBANK :http://www.drugbank.ca/
first go to this link enter your protein or condition like any disease and disorder it will give you complete information about the drug which is designed for the particular purpose eg enter anxiety disorder in drugbank it will give you different ligands of that particular disorder and protein and also gives complete information about the target protein.
The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. The database contains nearly 4800 drug entries including >1,480 FDA-approved small molecule drugs, 128 FDA-approved biotech (protein/peptide) drugs, 71 nutraceuticals and >3,200 experimental drugs. Additionally, more than 2,500 non-redundant protein (i.e. drug target) sequences are linked to these FDA approved drug entries. Each DrugCard entry contains more than 100 data fields with half of the information being devoted to drug/chemical data and the other half devoted to drug target or protein data.
Steps Involved in Drug Designing
3:15 AM
GCbioinfo
Drug designing steps were usually divided into steps as follows:
I. LIGAND PREPARATION
II. RECEPTOR PREPARATION
III. DOCKING
IV. BINDING AFFINITY STUDIES
I. LIGAND PREPARATION
II. RECEPTOR PREPARATION
III. DOCKING
IV. BINDING AFFINITY STUDIES
DRUG DESIGNING
3:13 AM
GCbioinfo
Drug designing basically of two types namely ligand based approach or receptor based approach. In both the case the point of centre only differ but requirement of receptor and ligand essential in both the case.
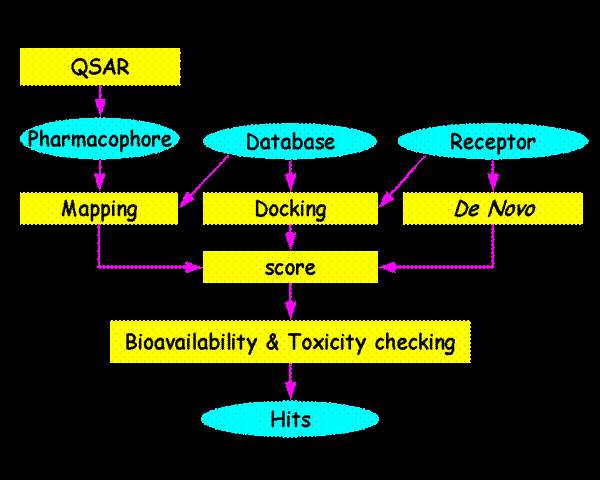
he shortcoming of traditional drug discovery; as well as the allure of a more deterministic approach to combating disease has led to the concept of "Rational drug design" (Kuntz 1992). Nobody could design a drug before knowing more about the disease or infectious process than past. For "rational" design, the first necessary step is the identification of a molecular target critical to a disease process or an infectious pathogen. Then the important prerequisite of "drug design" is the determination of the molecular structure of target, which makes sense of the word "rational".
In fact, the validity of "rational" or "structure-based"drug discovery rests largely on a high-resolution target structure of sufficient molecule detail to allow selectivity in the screening of compounds. Simple flowchart for drug designing shown in the figure:
Monday, October 11, 2010
HTML Frames:
10:59 AM
GCbioinfo
I told you people about the html frames there is the same thing but with the slight variation
<html>
<frameset rows="20%,80%">
<frame src="file:///E:/anny.html" name="frame1" noresize="noresize">
<frame src="http://www.islamickorner.com">
</frameset>
</html>
one new thing is noresize attribute which means you can not resize your page as previously by dragging its size is fixed you can check it by dragging your page it can't be happened.
the out put is as:

<html>
<frameset rows="20%,80%">
<frame src="file:///E:/anny.html" name="frame1" noresize="noresize">
<frame src="http://www.islamickorner.com">
</frameset>
</html>
one new thing is noresize attribute which means you can not resize your page as previously by dragging its size is fixed you can check it by dragging your page it can't be happened.
the out put is as:

HTML Images2:
10:42 AM
GCbioinfo
I told you how to link your page to other sites by using the <a href> tag so I have an assignment for you design a page in which you click on image to access the new page instead of a text eg you click Islamickorner to access this site but now click an image to access this site so try it
out put is as:
click the second image to get the next page
 this is the next page or site which is accessed by the link behind that image its code is as:
this is the next page or site which is accessed by the link behind that image its code is as:
<a href="http://www.bermuda-online.org/roses2.gif"><img src="file:///C:/Documents%20and%20Settings/Administrator/My%20Documents/My%20Pictures/roses2.gif" height="200" width="200" border="10"></img></a>
out put is as:

click the second image to get the next page
 this is the next page or site which is accessed by the link behind that image its code is as:
this is the next page or site which is accessed by the link behind that image its code is as:<a href="http://www.bermuda-online.org/roses2.gif"><img src="file:///C:/Documents%20and%20Settings/Administrator/My%20Documents/My%20Pictures/roses2.gif" height="200" width="200" border="10"></img></a>